Eine Genfamilie ist eine Sammlung von Genen, die durch Gen- oder Genom-Duplikationsereignisse aus einem einzigen Gen entstand, ähnliche Nukleotid- oder Proteinsequenzen hat und in der Regel ähnliche Funktionen ausführt. So oder so ähnlich lautet die Definition, die hoffentlich alle Biologiestudierenden gelernt haben. In der Biologie ist dies eine ziemlich eindeutige Definition, wenn man bedenkt, dass die Definition des "Gens" noch immer umstritten ist.
Aufmerksame Leser*innen werden das Thema dieser Planter‘s Punch-Ausgabe erraten haben. In dieser Ausgabe ist ein "Gen" die kontinuierliche DNA-Sequenz zwischen dem Anfang und dem Ende eines Gens. Und schließlich möchte ich erklären, warum die Analyse von Pflanzengenfamilien wichtig ist. Die Analyse und der Vergleich von Pflanzengenfamilien kann grundlegende wissenschaftliche Fragen lösen, z.B. wie sich Pflanzen diversifizieren, und kann gleichzeitig in der Praxis Anwendung finden, z.B. um die Vollständigkeit eines neu sequenzierten Genoms zu beurteilen. Die Genomsequenzierung ist der Prozess der Entschlüsselung und Bestimmung der DNA-Sequenz eines Organismus. Pflanzen diversifizieren, indem sich in ihrem Erbgut, d.h. ihrem Genom, Änderungen akkumulieren. Je mehr von diesen Änderungen stattfinden, umso mehr unterscheiden sie sich genetisch von Ihrem Vorgänger. Diese Veränderungen können die Überlebensfähigkeit einer Pflanzenart verbessern und es ihr ermöglichen, in einer anderen oder sich verändernden Umgebung zu gedeihen, z. B. aufgrund des Klimawandels oder durch den Erwerb einer Resistenz gegen einen Pflanzenschädling.
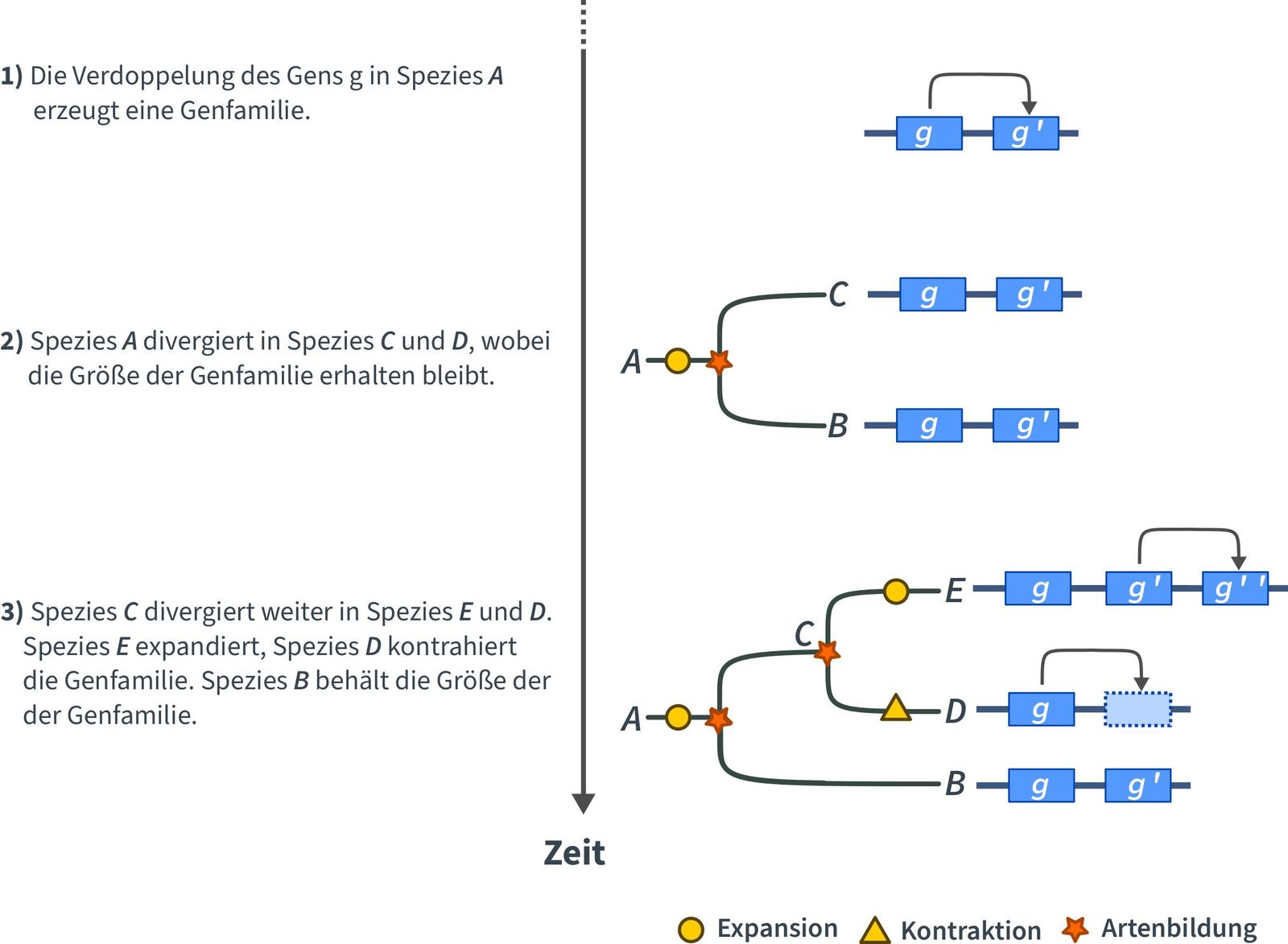
Die Duplikation oder Deletion, d.h. der Verlust eines Gens, kann einen starken Einfluss auf die Fitness und Evolution einer Pflanze haben. Die Auswirkung einer Duplikation oder Deletion hängt von der Bedeutung des betreffenden Gens und seiner Rolle in den Stoffwechselvorgängen ab, an denen es beteiligt ist, bzw. war. Wenn ein Gen dupliziert wird, expandiert die Genfamilie, d.h. sie wächst. Geht ein Gen verloren, kontrahiert die Genfamilie, d. h. sie schrumpft. Die Größe einer Genfamilie variiert und kann von zwei Mitgliedern bis zu mehr als hundert reichen. Darüber hinaus kann die Größe einer Genfamilie zwischen zwei Arten ebenfalls variieren. Wir gehen jedoch davon aus, dass die Größe einer Genfamilie, die an grundlegenden und wichtigen Stoffwechselvorgängen beteiligt ist, sehr ähnlich ist.
Die Expansion oder Kontraktion einzelner Genfamilien innerhalb eines Pflanzenstammbaums erfolgt zufällig oder ist das Ergebnis eines natürlichen Prozesses, d. h. eine Veränderung in einer Genfamilie hat einer Pflanze in irgendeiner Weise genutzt. Die Analyse von Genfamilien hilft, solche Ereignisse und den anschließenden Einfluss auf die Pflanzenevolution zu identifizieren. Die Einbeziehung von ferner verwandten Pflanzenarten in solche Studien, z.B. Arabidopsis thaliana, Triticum aestivum (Brotweizen), Moose, Algen, kann helfen, den Zeitpunkt der Veränderung zu bestimmen.
Bevor ein Vergleich durchgeführt werden kann, müssen Genfamilien definiert und identifiziert werden. Beide Schritte werden in der Regel auf Proteinebene mit Hilfe von Proteinsequenzen durchgeführt. Proteine sind Aminosäuren, welche miteinander zu einer Kette verbunden sind. Ein Gen kodiert die einzelnen Aminosäuren mittels spezifischer Codons für jede Aminosäure, und die Reihenfolge dieser Codons in einem Gen kodieren das Protein. Zum Teil hat eine einzelne Aminosäure mehrere Gencodes, d.h. unterschiedliche Gencodes codieren die gleiche Aminosäure. Dies nennt man die Codon Degeneration, bzw. Redundanz, des genetischen Codes. Daher können Proteinsequenz zwischen verschiedenen Spezies ähnlicher ist als der unterliegende Gencode. Dies vereinfacht die Identifikation von Sequenzen mit ähnlichen Funktionen aus weiter auseinander liegenden Sequenzen.
Um eine Genfamilie zu definieren, werden bekannte Proteine mit ähnlichen Sequenzen zu Proteinclustern gruppiert, so dass jedem Cluster eine biologische Funktion zugeordnet werden kann. Schließlich wird jeder spezifische Cluster mit einem Modell beschrieben, das verwendet werden kann, um bestehende oder neue Genome nach bestimmten Genfamilienmitgliedern zu durchsuchen.
Die einzelnen Modelle werden manuell überprüft und kontinuierlich aktualisiert. Außerdem lassen sich mit diesen Modellen sehr schnell Genfamilien identifizieren. Das Proteom, d.h. alle bekannten Proteine einer Pflanzenspezies, kann in wenigen Minuten analysiert werden.
Die aktuelle und stetig wachsende Fülle an verfügbaren Genomdaten erlaubt es uns nun, Vergleiche zwischen zahlreichen, zum Teil sehr unterschiedlichen Pflanzenarten durchzuführen. Zu diesem Zweck setzen wir Hochdurchsatzverfahren ein, um die Proteome dieser Pflanzen zu screenen. Damit können wir die Expansion und Kontraktionen innerhalb bekannter Genfamilien identifizieren, um artspezifische Genfamilien zu ermitteln. Darüber hinaus versuchen wir herauszufinden, wann diese wichtigen Ereignisse während der Evolution der verschiedenen Pflanzenarten aufgetreten sind.
Durch die Codondegenerierung erleichtern Proteinsequenzen die Identifizierung und den Vergleich von Genfamilien zwischen verschiedenen Pflanzenarten. Außerdem sind sie in großen Mengen verfügbar. Ebenfalls sind bewahrte und gründlich getestet Werkzeuge und Methoden zum Umgang mit diesen Daten verfügbar. Proteine sind keine linearen Konstrukte, sondern falten sich zu charakteristischen Strukturen und einzelne Strukturen können miteinander interagieren, um größere und komplexere Strukturen zu bilden. Diese Strukturen bestimmen die Funktion eines Proteins, zum Beispiel eines Enzyms. Bemerkenswerterweise können sehr unterschiedliche Proteinsequenzen zu ähnlichen Strukturen führen, die ähnliche Funktionen erfüllen.
Es stellt sich die Frage, warum wir dann immer noch Proteinsequenzen verwenden, um Genfamilien zu identifizieren? Ein Haupthindernis ist bisher das Entschlüssen von Proteinstrukturen, da dieser Prozess experimentell noch sehr komplex und langwierig ist. Allerdings haben Wissenschaftler*innen und Ingenieur*innen an Hochschulen und Unternehmen in letzter Zeit große Fortschritte bei der Entwicklung von Software zur Vorhersage von Proteinstrukturen gemacht. Leider erfordern diese Ansätze immer noch riesige Mengen an Computerleistung und Hardware. Allerdings wird dies nicht die Wunderlösung sein, um Genfamilien zu untersuchen. Ähnliche Proteinstrukturen zwischen Spezies können von einem ursprünglichen Gen abstammen. Es kann aber auch sein, dass ähnliche Strukturen unabhängig voneinander evolviert sind, d.h. nicht von einem gemeinsamen Gen, sondern von einem oder mehreren, nicht verwandten Genen. Diesen Vorgang nennt man konvergente Evolution. Beispiele konvergenter Evolution in Pflanzen ist die C4-Photosynthese und die Evolution zu Fleischfressenden Pflanzen. Diese Eigenschaften entstanden nicht einmal, sondern mehrere Male in verschiedenen Pflanzenfamilien. Ein anderes Beispiel ist die die Evolution des Auges zwischen Wirbeltieren (also auch uns Menschen), Kopffüßlern, (z.b. Kraken), und Nesseltieren, welches sehr ähnlich aussieht und funktioniert, aber in jeder dieser Gruppen unabhängig voneinander entstanden ist. Konvergente Evolution muss dann bei Vergleichen von Proteinstrukturen miteinbezogen und analysiert werden, z.B. mit der Analyse von Genfamilien.
Mittel Proteinsequenzen können wir nicht sehr weit entfernte Mitglieder eine Genfamilie identifizieren, aber konvergente Evolution beeinflusst unsere Analysen nicht so fest. Andererseits könnten wir Genfamilien mittels Proteinstrukturen von sehr unterschiedlichen Spezies untersuchen, müssen dann aber zusätzliche evolutionäre Pfade stärker berücksichtigen.
Aber bis wir Genfamilien mit Strukturen in Pflanzen untersuchen können, muss man mehr pflanzliche Proteinstrukturen experimentell entschlüsseln oder vorhersagen und testen können. Bis dahin sind wir auf die Verwendung von Proteinsequenzen angewiesen.
Planter’s Punch
Unter der Rubrik Planter’s Punch wird jeden Monat ein bestimmter Aspekt des CEPLAS-Forschungsprogramms vorgestellt. Alle Beiträge werden von Mitgliedern der Graduiertenschule und des Postdoc-Programms erstellt.

Über den Autor
Jan Buchmann ist Postdoc in der Gruppe von Prof. Björn Usadel im Institut für Biological Data Science an der Heinrich-Heine-Universität Düsseldorf. Seine Forschungsschwerpunkte sind die Evolution von Genfamilien in Pflanzen und die Entwicklung von Methoden zur Identifizierung von pflanzlichen Gen-Unterfamilien in Genomen ohne vorheriges Gen-Calling. Er promovierte an der Universität Zürich in der Schweiz und analysierte den Einfluss von Transposons auf die Evolution von Pflanzengenomen. Bevor er zu CEPLAS kam, untersuchte er die Genomevolution von Viren und entwickelte Methoden zur Identifizierung neuer Viren in metagenomischen Datensätzen an der Universität von Sydney in Australien.